
Traditional practice for treating a broken tibia shaft utilizes a central nail with load-sharing capabilities to produce high stability and early postoperative patient mobilization, referenced as EXPERT NAIL™ Tibial Nails. In a recent review of this treatment, it was determined that several outcome-related problems were identified for the purpose of engineering a new treatment method attempting to address them. These issues are comprised of delayed bone union, malunion, nonunion, patient pain and discomfort, anatomical nail fit, surgeon usability aspects, and instrumentation for various surgical approaches.
A clinical solution, given the name TN-Advanced Tibial Nail (TN-A) incorporates five proximal and four distal multiplanar locking capabilities with an added design feature for the purpose of providing angular stability. This innovative nailing system is specifically designed to enable surgeons to correct shorter bone fragments with elevated stability for the purpose of elevating bone healing and avoid secondary loss of reduction. One significant advantage of this new design is wherever the locking screws are inserted into the locking holes of the TN-A (except the proximal 7 mm nail slot), the inlays offer angular stability between locking screws and nail without any need for added instrumentation and surgical steps. The optimal candidates for the TN-A implants are adults and adolescents ranging from 12 – 21 years of age, where growth plates have already fused together. More specifically, these implants are for open and closed proximal and distal tibial fractures, open and closed tibial shaft fractures, and tibial malunions and nonunions.
Regulatory and quality standards for temporary implantable devices (class III) such as the TN-A are extensively detailed, precise and highly intricate. EMMA International has incurred specialized firsthand experience implementing a streamlined Quality Management System for successful Class III medical device quality compliance and FDA regulatory submissions.
As the world opens back up, EMMA International is here to provide full-circle solutions for all aspects of the MedTech industry. Give us a call at 248-987-4497 or email us at info@emmainternational.com to learn more about how EMMA International can take the stress out of quality and regulatory compliance!
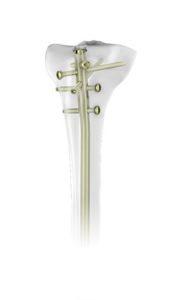
Approved Solutions (2020) TN-Advanced Tibial Nailing System, Retrieved on 06/02/2021 from https://approvedsolutions.aofoundation.org/approvedsolutionsfolder/2020/tn-advanced-tibial-nailing-system#tab=details;



