
Every year FDA releases an annual report for Quality system inspections and enforcement action data for the Medical Device industry. For our blog this week, we are presenting a brief analysis of the data published by the FDA for the fiscal years 2015, 2016 and 2017 prior to the release of the FY 2018 data in December. Here are some of our key observations from the current data.
In 2015, 2016 and 2017, FDA conducted 2271, 2246 and 2235 inspections respectively.* The number of inspections has remained fairly consistent; however, the breakdown of the number of domestic and international sites is noteworthy.
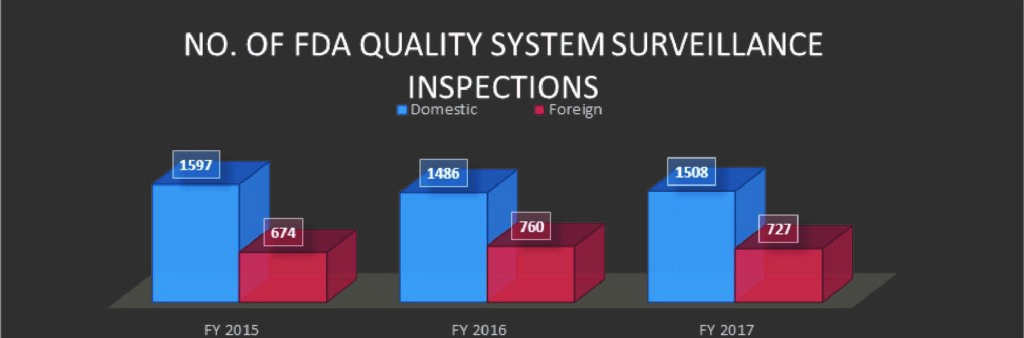 FIG A*
FIG A*
The number of International sites inspected by the FDA increased from 674 in 2015 to 760 and 727 in 2016 and 2017 respectively, which could be an indication of more and more foreign manufactured products entering the US market or US companies outsourcing their manufacturing, thereby requiring the need of FDA inspecting the said international sites. It is worth noting that there is a gap in the literature, and any applicable publications, surrounding any explanation behind the number of domestic or foreign sites’ inspections.
The number of 483s issued to medical device companies for quality system violations showed an increase of only 14% from 2016 to 2017 in comparison to the drastic decline in the number of warning letters issued.
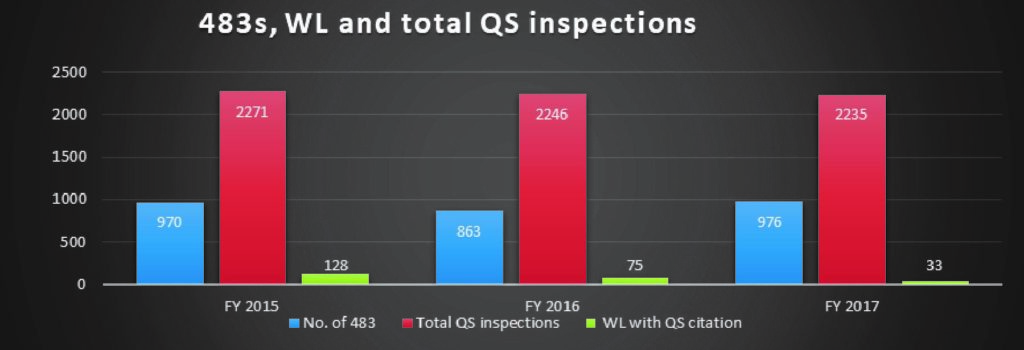 FIG B*
FIG B*
There was a 56% decline in the number of warning letters issued between FY 2016 and 2017, and a 41.4% drop between the FY 2015 and 2016.
Let’s take a peek at the trends in the QSR subsystem citations in the 483s and warning letters.
CAPA and Production and Process Controls continue to remain the most frequently cited observations in all three years.
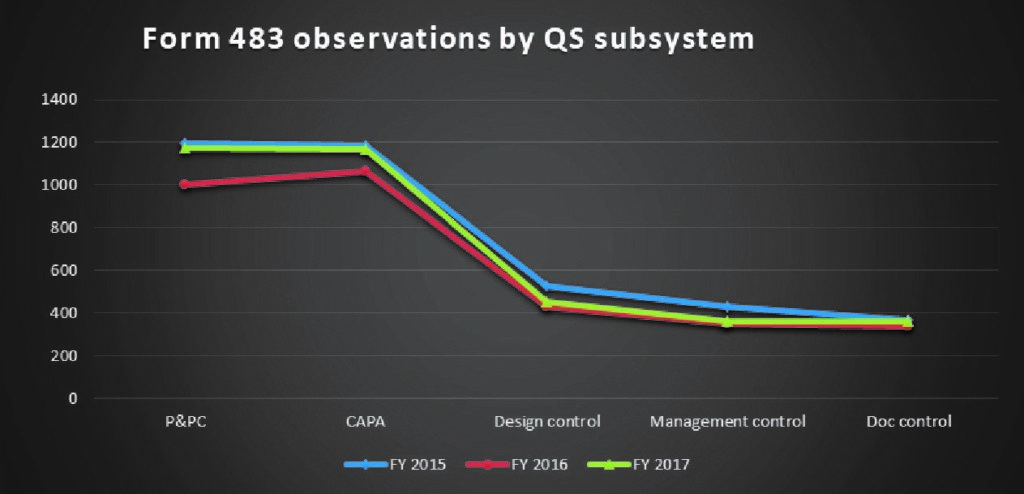 FIG C*
FIG C*
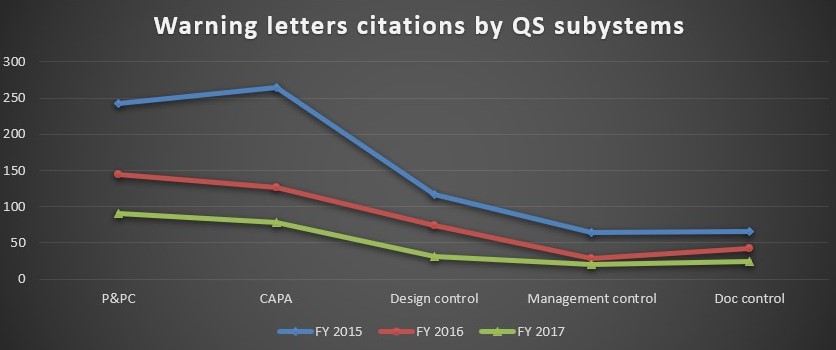 FIG D*
FIG D*
Fig E. below shows the share of CAPA and P&PC citations in warning letters issued since 2015. CAPA and Production and Process Controls appeared in more than 50% of the total warning letters each year.
The other citations in the warning letters include violations in design control, management control, and document control.
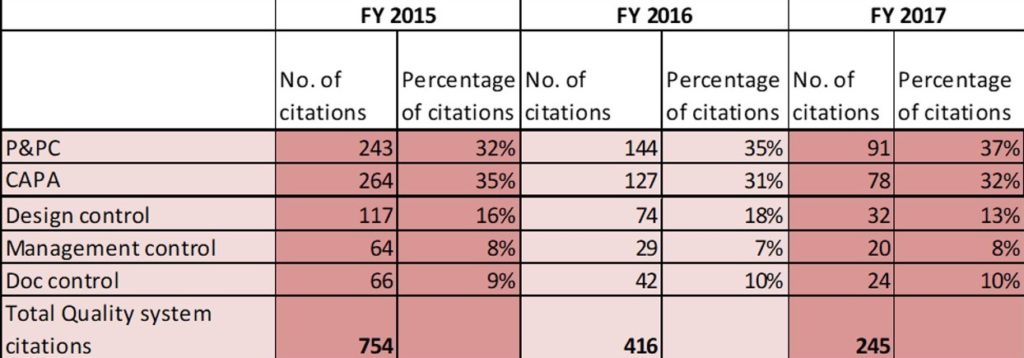 FIG E*
FIG E*
Fresh data (for FY 2018) is soon to be released, but until then we will have to wait to see what new trends would highlight. We will monitor the trends and keep you updated on what we find out from the new 2018 data.
If your company needs help with Quality System remediation, 483 or warning letter responses, please call (248) 987-4497 or send an email to info@emmainternational.com.
*Data Source: FDA (Dec 2017) FY2017 Annual FDA Medical Device Quality System Data retrieved on 11-16-2018 from https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/UCM597261.pdf



